


PHARMA PRODUCT DEVELOPMENT AND SUPPLY CHAINS
Pharma doesn’t follow other industries

[Note: This is taken from COVID Supply Chains: Fact not Fiction]
DISCOVERY RESEARCH ‘FINDS’ A MOLECULE
In most industries, supply chains are created as part of a new product development program. The developers work together, consulting product end-users, producers, and distributors to ensure the physical supply chain they put together can deliver what is required by consumers.
For reasons we shall go into later, pharmaceutical new product development does not follow the customary approach of other industries. Rather than beginning with a consumer and working upstream to the beginning, the process begins with a patented compound and moves forwards.
This results in supply chains ‘evolving’ with the passage of time, rather being designed and planned with the consumer in mind.
The ‘R’ of R&D (Discovery Research) discovers (or finds) molecules using advanced technologies such as molecular modelling. Once it is confirmed a patent is in place, promising compounds are handed to ‘D’ (Development).
PRODUCT DEVELOPMENT HAS TO FILE IT WITH THE REGULATORS
Development is a completely new team of specialists responsible for building a supply chain for pre-clinical testing initially. That means proving the test compound that will be produced by the supply chain is safe to study in humans. For small molecule products (made by industrial chemistry), around 5 – 10 kilograms of compound will be produced.
Figure 5 shows a typical small molecule product supply chain for the production of the active pharmaceutical ingredient (API).
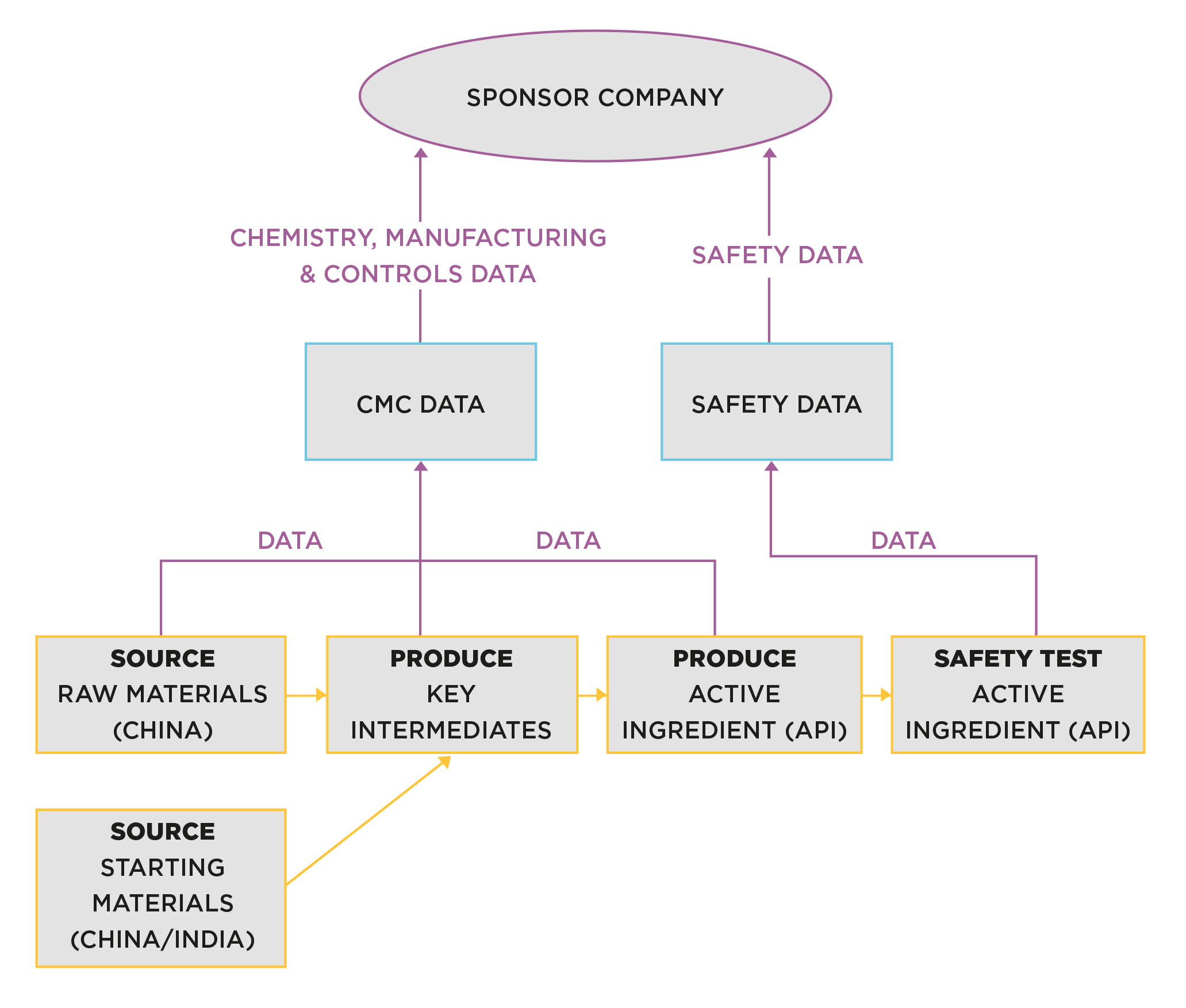
Figure 5. Production of API for preclinical testing
The production process is devised by a ‘route scout’ (process chemist). Their job is to convert a laboratory process that had only ever produced small gram quantities, into a process that can yield much larger quantities. The skill involved is about making the process as efficient and ‘manufacturable’ as possible.
A small-scale pilot plant is normally used at this stage, as the volume is not sufficient to justify occupation of commercial-scale plant. Figure 5 shows
Sources of raw and starting materials will be selected by the scientists working on the project. In this case, there are intermediate chemicals involved. Intermediate stages are not always required; it depends on the chosen route of synthesis.
Once produced, the API is transported for safety testing at one or more chosen Contract Research Organisations (CROs).
Even though this appears to be a simple supply chain, there is already an array of suppliers and service providers involved, often spanning the globe. Remember, raw, starting materials and intermediates are sourced primarily from China and India, so ex-Asian countries are operating a long way from home.
For Western companies, this puts a limit on due diligence in selecting sources, and oversight when selected sites are in operation. It also adds significantly to lead-time and complexity.
FROM SAFETY TESTING TO STUDIES IN HUMANS
If it looks like the results support moving to studies in humans, the company will fill out and submit an Investigational New Drug application (IND), a Clinical Trial Application (CTA) in UK/EU.
The IND must include all the details about the supply chain and safety data. If approved by the Regulatory Authority (RA), the company will be awarded an IND and is designated a clinical trial sponsor (CTS).
The CTS will plan to produce product for the first stage of clinical trials - phase I studies in healthy volunteers. The compound being studied is known as ‘test material’.
This is the beginning of an application to market the product, known as a New Drug Application (NDA) or Biological License Application (BLA) in the US, and Marketing Authorisation Application (MAA) in UK/EU.
The programme of work that needs to be carried out by the CTS to prove quality, safety and efficacy is laid down by the RA in the Common Technical Document (eCTD). The details of the end-to-end supply chain have to be specified in Module 3 of the eCTD.
THE COMMON TECHNICAL DOCUMENT (eCTD)
The eCTD is the template that must be used to submit a licence application to market a drug. This format has been agreed by RAs globally following regulatory collaboration and the work of the International council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Below is the agreed pyramid structure providing an overview of what is required:
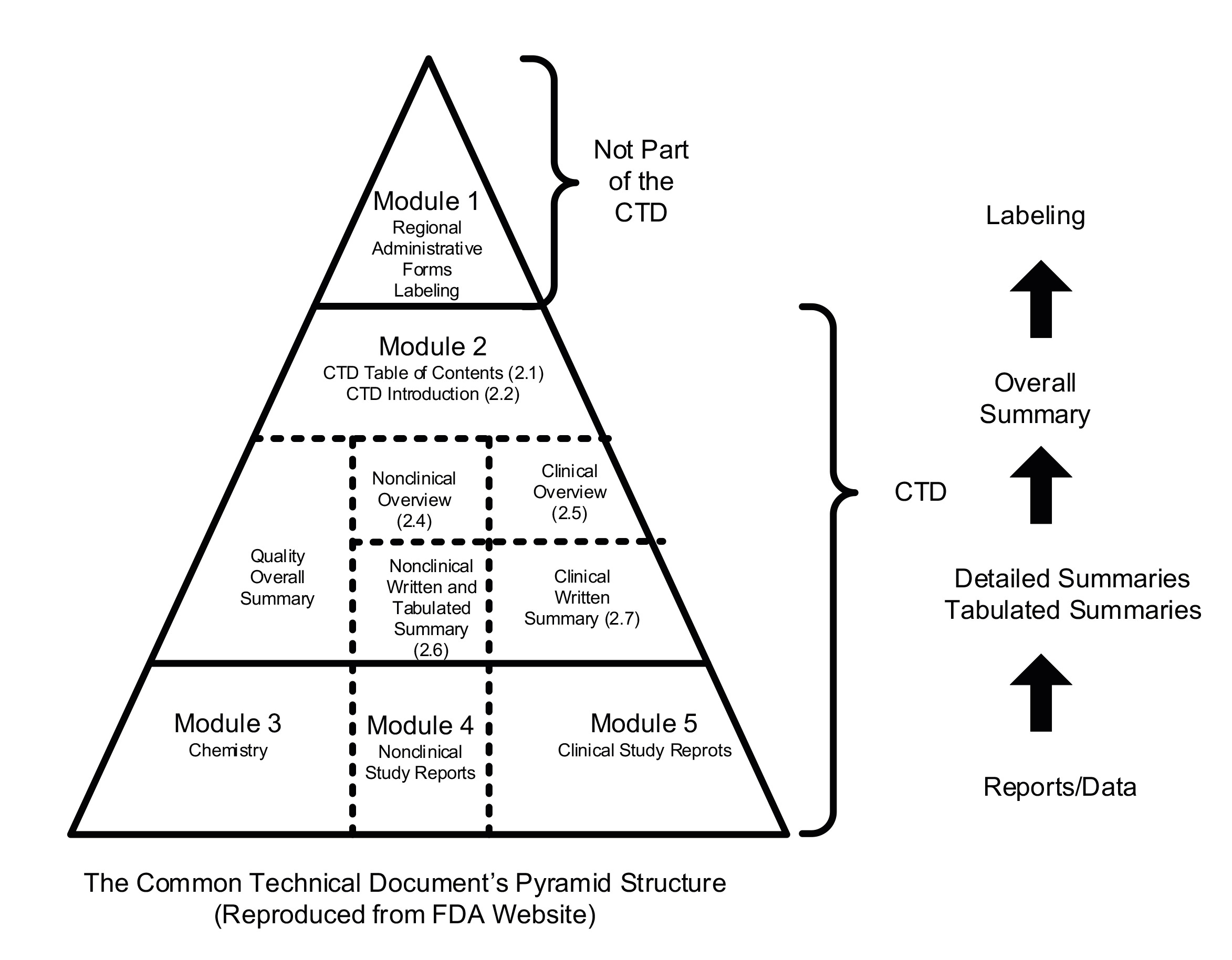
Figure 6. Common Technical Document (electronic)
The three modules are at bottom of the pyramid:
Module 3: Chemistry, manufacturing & controls (CMC or Quality)
Module 4: Nonclinical Study reports
Module 5: Clinical Study Reports.
The sections above the modules provide overviews and summaries.
Module 3 is titled ‘Chemistry’. This is short for ‘chemistry, manufacturing, and controls’ (CMC). The CMC section of the dossier is where all the details about suppliers, manufacturers, material and product specifications, test procedures, etc must be declared.
To gain approval from FDA, the production facilities making the active pharmaceutical ingredient (API) and drug product (DP) must undergo a thorough physical inspection by suitably qualified inspectors. These are known as pre-approval inspections (PAIs).
There is an amount of regulatory flexibility when it comes to standards in the supply-chain for producing preclinical test material, given the early stage of development. For trials in humans, however, production in the supply-chain must comply with Good Manufacturing Practice (GMP) and Good Distribution Practice (GDP).
The clinical trial supply chain, Figure 7, shows the extension of the supply-chain stages into production of the dosage form and clinical trial kits, to be shipped into storage awaiting call-off from study site locations.
Each additional stage is likely to be in different parts of the world, with a number of different service providers at each stage.

Figure 7. Production clinical trial supply chain
When the time comes to submit the regulatory filing (NDA/BLA/MAA, the Regulatory Authority mandates that all data are collected and submitted using the eCTD format.
WHO WAS THINKING ABOUT THE SUPPLY CHAIN TO MARKET?
You may be surprised to learn that no-one is thinking about the supply chain that will be required to support a market launch—all the thought and effort went into collecting the data required by the RA to award an approval.
Food for thought?
If they make it, doctors will prescribe it and patients will willingly use it.
They are a law to themselves, they don’t give a damn.