

FDA Notification of FOI Expedite Request Denied Control # 2024-5142
FDA Notification of FOI Expedite Request Denied Control # 2024-5142
I have appealed to the Office of the Executive Secretariat, US Food & Drug Administration

The title is from an email from FDA earlier today:
Expedite Request Denied Control # 2024-5142
This is my appeal letter, just sent:

Office of the Executive Secretariat
US Food & Drug Administration
5630 Fishers Lane, Room 1050
Rockville, MD 20857
FDA FOIA reference file: 2024-5142
Dear Office of the Executive Secretariat,
I am in contact with Ms. Sly in relation to the FOIA reference above. The information provided was not initially as requested, but during a telephone conversation with Ms. Sly, June 17, 2024, I agreed to accept a previously released Establishment Inspection Report (EIR) for the CBER pre-license inspection ending July 23, 2021, at the Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC (used to support the original approval of COMRINATY, BLA 125742) in fulfillment of CBERs portion of my request.
The support I received from Ms. Sly was excellent. However, I was puzzled as to why the pre-license inspection above was not on the FDA inspections database, shown below (accessed 05:20, 06/21/2024).
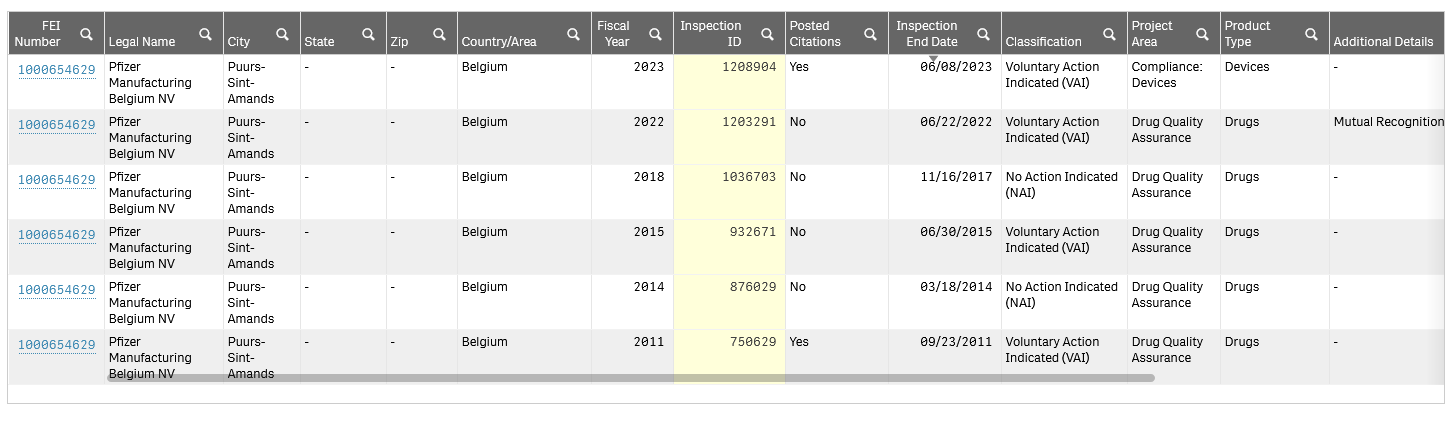
This meant that I was not able to ascertain the regulatory classification of the inspection. I felt it was crucially important to know the classification, as the OBJECTIONABLE CONDITIONS AND MANAGEMENT RESPONSE: Observations listed on form FDA 483 (EIR, page 60 onwards) were substantial, as recounted in my email to Ms. Fry, 06/19/2024, in the excerpt from APPENDIX I:
“The 13 observations relating to cGMP non-compliances indicate (to me at least) serious deficiencies within the Wyeth Biopharma facility, especially in relation to the potential for microbial, particulate and pyrogen contamination. If I may direct you to the CFR Title 21 link: 211.113 Control of microbiological contamination:
(b) Appropriate written procedures, designed to prevent microbiological contamination of drug products purporting to be sterile, shall be established and followed. Such procedures shall include validation of all aseptic and sterilization processes.”
With a 40+ year career in biopharmaceutical manufacture and distribution, I find it difficult to understand how the Wyeth facility could have been licensed for the manufacture of a drug substance (DS) for a sterile injectable product to be placed into interstate commerce. The EIR speaks to systemic failure of the Wyeth quality system that would indicate significant remediation work had to be carried out prior to resumption of production. This is why I requested Expedited Processing for FOIA 2024-5142, according to this entry in the form submitted:
“The EUA process did not make provision for the sufficient review of Module 3 – Chemistry, Manufacturing & Controls (CMC) of the electronic Common Technical Document (eCTD), to ensure safety of patients. I would like to draw FDAs attention to the heparin incident of 2007-8, where economic adulteration of a constituent material used in manufacture led to significant patient harm, please see: https://www.pewtrusts.org/-/media/legacy/uploadedfiles/wwwpewtrustsorg/reports/health/pewheparinfinalhrpdf.pdf. I believe the [apparent] gap in biologics inspection history (from April 2017 to September 2023) at the Wyeth Biopharma facility, would have prevented FDA from identifying critical non-compliances that would have been a danger to human life.”
In further support of regulatory action being considered for COMRINATY, BLA 125742, the FDA Inspection history for the drug product (DP) manufacturer, Pfizer Manufacturing Belgium NV, FEI Number 1000654629, is mainly made up of refusals, with none for biologics accepted, as shown below:
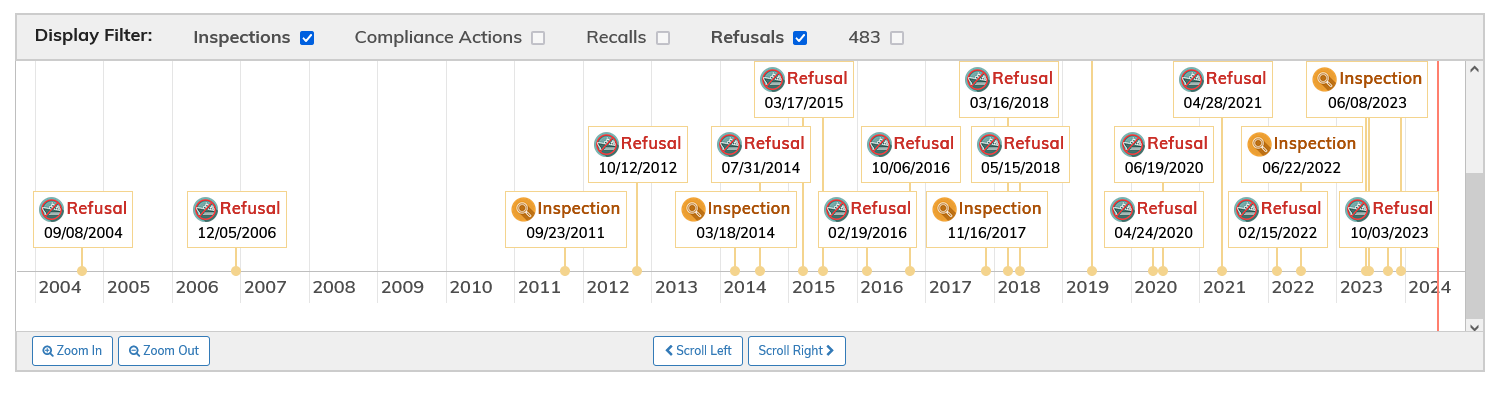
The implication is (to be confirmed) that FDA did not carry out a biologics-grade inspection on this facility. I can see there is an inspection listed as: mutual recognition. However, given the drug product (DP) is a sterile injectable, it would seem essential that FDA protect US consumers with a physical inspection, rather than rely on a third-party agency.
Additionally, there is this a publicly available Form 483 for another Comirnaty manufacturer Rentschler Biopharma: Rentschler slapped with FDA Form 483 citing lax manufacturing procedures. To my knowledge, no regulatory action has been taken up to this point.
The concluding matter relates to FDAs oversight of the COMRINATY temperature sensitive supply chain from starting materials through upstream and downstream processing into DP. It has been my experience that USP <1079> (Good Storage and Distribution Practices) is often used by FDA as a guide to good practice in the manufacturing and distribution supply chain. By my understanding, the temperature ranges for products and materials in the SARS-CoV-2 sterile injectables supply chain included at least:
· +2℃ to +8℃
· +15℃ to +25℃
· -25℃ to -15℃,
· -60℃ to -40℃,
· -80℃ to -60℃.
· -190 °C to -150 °C
This raises the question as to how the FDA was able to ensure that any temperature excursions identified at any stage in the supply chain were properly investigated and corrective and preventative action (CAPA) implemented?
In consideration of the above, whilst I understand it may not be wholly within the bounds of a FOIA request, I would ask that these issues be passed through to FDA executive management for urgent consideration of the serious issues relating to US consumer safety.
Your kind response would be appreciated.
Sincerely,
Hedley
Hedley Rees
Managing Consultant and Author
Cell: +44 7734 961726
Book: Supply Chain Management in the Drug Industry: Delivering Patient Value for Pharmaceuticals and Biologics - Read Me
APPENDIX I: Text of email to Ms. Sly, dated 06/19/2024
Dear Ms. Fry,
I appreciated your call and the information you provided. You kindly offered to guide me through the various FDA processes and that, again, is very welcome. You made the point that an FOIA provides information requested, but questions cannot be asked; also, that further FOIAs may incur costs that could be significant. I have very little budget so will need to tread carefully on that. Although I operate a consultancy in biopharmaceutical supply chain management, I have had no clients since the beginning of COVID.
If I could first ask for some clarifications on Wyeth BioPharma inspections and the EIR provided, which ended on 07/23/2021. I was not able to find it on the inspections dashboard: https://datadashboard.fda.gov/ora/firmprofile.htm?FEIi=1222181&/identity/1222181 . Have I performed the query incorrectly, or is there some other reason I do not understand relating to its omission? It would be useful to know if the regulatory classification was NAI, OAI or VAI?
Also, should I take it that the information requested in FOIA Control # 2024-5142, on Inspection ID: 1020234 and Inspection ID: 1223617, would be the subject of further FOIAs that may incur significant cost?
I have now read through OBJECTIONABLE CONDITIONS AND MANAGEMENT RESPONSE: Observations listed on form FDA 483 (EIR, page 60 onwards).
If I may be allowed to make my own observation here and please do let me know if this is inappropriate within FDA procedure and process. The 13 observations relating to cGMP non-compliances indicate (to me at least) serious deficiencies within the Wyeth Biopharma facility, especially in relation to the potential for microbial, particulate and pyrogen contamination. If I may direct you to the CFR Title 21 link: 211.113 Control of microbiological contamination:
(b) Appropriate written procedures, designed to prevent microbiological contamination of drug products purporting to be sterile, shall be established and followed. Such procedures shall include validation of all aseptic and sterilization processes.
Additionally, given the complexity of the information systems required within the Wyeth Biopharma and the need to validate systems impacting cGMP, I wondered if the following section of CFR Title 21 was covered in the investigation?
TITLE 21--FOOD AND DRUGS: CHAPTER I--FOOD AND DRUG ADMINISTRATION, DEPARTMENT OF HEALTH AND HUMAN SERVICES, SUBCHAPTER A – GENERAL
PART 11 ELECTRONIC RECORDS; ELECTRONIC SIGNATURES
I look forward to working with you further on this, Ms. Sly, and would welcome any signposting required so that the questions I raise can be further considered within FDA, for possible regulatory action.
Sincerely,
Hedley
Hedley Rees
Managing Consultant and Author
PharmaFlow Ltd
T: +44 1656 655664
M: +44 7734 961726
Book: Supply Chain Management in the Drug Industry: Delivering Patient Value for Pharmaceuticals and Biologics - Read Me
What do you make of that? Is the FDA playing cat and mouse?
A noble effort! Most impressive paper trail....Thank you so much.
Thank you. Your rattling their cage! Great letters.
Blessings