


MHRAs Point of Care manufacture is complete madness—
Check in to find out why

Letter from MHRAs Dr Laura Squire
You will note from the screenshot below that I had pre-submitted a question to MHRAs March Board Meeting:

This is the full letter here:
Letter on Point of Care Manufacture.
This is Dr. Laura Squire OBE.
Her responsibilities:
“Laura oversees a large portfolio that is designed to ensure the quality and access of products to the UK market - this includes scientific advice, clinical trials/clinical investigations, licensing assessment, marketing authorisations and device registrations, inspections, enforcement and standard setting through for example the British Pharmacopoeia and Target Product Profiles.” Oh, and she locks up the MHRA offices every night before she goes to bed 😊!
Her qualifications:
“She joined the Medicines and Healthcare products Regulatory Agency from the Department of Health and Social Care, where she worked extensively on the COVID-19 vaccine deployment programme. Well qualified then!”
This is serious, very serious
It’s difficult to overstate the seriousness of this. The letter says:
“The proposed mechanism for oversight is a PoC Master File that names the ‘Control site’ and all satellite sites that carry out manufacture of PoC products.”
Then it goes on:
“The Control site must hold an appropriate licence (e.g. MIA or (MIA)IMP) and would be responsible for all satellite sites.”
So, where are the Control Sites? Who will be in charge? How many satellite sites will there be? Presumably they are hospital pharmacies, if it is Point of Care manufacture?
Where skills experience and quality systems will they employ?
You may remember this previous newsletter:
MHRA confirms that hospital pharmacies can manufacture, transport and store gene therapy products, and the Care Quality Commission will be responsible, not MHRA

Delighted to report there has been a breakthrough with MHRA This morning, I received this email from Ian Rees, Manager, Inspectorate Strategy and Innovation Unit” “Dear Hedley Many thanks, I had a good break. Thank you for your reply and I can confirm that I have passed this information on to the Directorate.
This is what I warned in that Newsletter:
“The answer below is very helpful, albeit it does not address the majority of the questions posed [in the FOI]. In spite of that, it has allowed me to establish that any ATMP approved by MHRA under the changed regulations, has the potential to be misbranded. The logic is this. The application from a clinical trial sponsor (CTS) to market an ATMP must follow the MHRA Marketing Authorisation Application (MAA) process on the MHRA website, which says: You should submit your application using the electronic Common Technical Document (eCTD). Since this is an open letter, as well as a FOI request, I will explain the eCTD process below. This is meant for the public at large, and I am not suggesting for one minute that you do not know the process.
When a company applies to MHRA to market a new medicinal product, it is required to prepare and submit what is known as an electronic Common Technical Document (eCTD). The eCTD format has been agreed and adopted globally, with every country providing the same data and information required to evaluate a medicinal product for sale. The global template was produced under the auspices of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The pyramid structure below shows, at the bottom, the three modules that must be submitted:
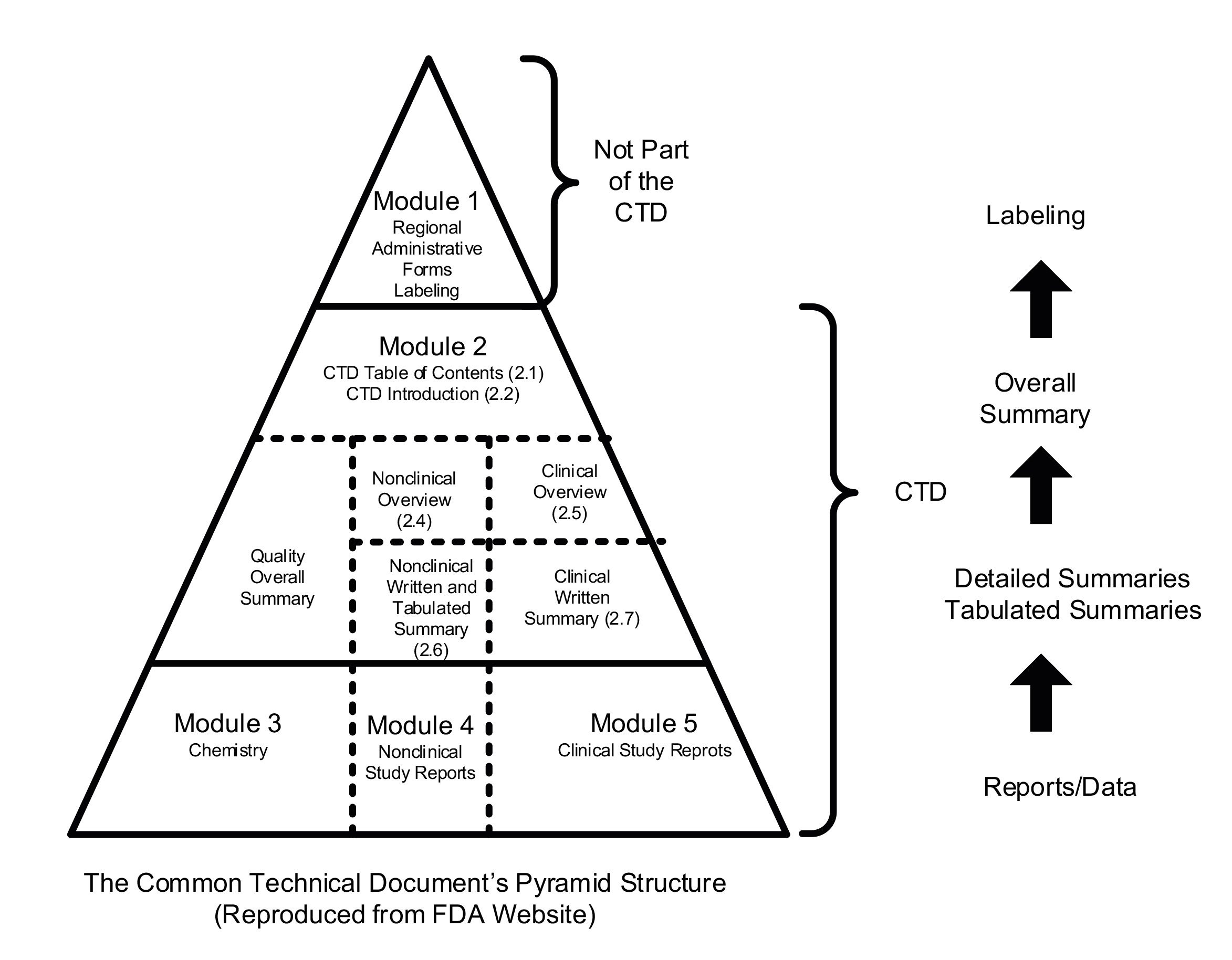
Module 3: Chemistry (full name is Chemistry, Manufacturing and Controls (CMC) or Quality Module).
Module 4: Nonclinical (Safety, Toxicology etc of manufactured product).
Module 5: Clinical (efficacy).
Module 3 is critical to patient safety, as it represents the entire supply chain that will produce the product to be administered to the patients who will receive it. In Module 3, every stage of manufacture, storage and transportation must be described in immense detail. The same applies to determine the methods to test the quality of the product at key stages, until it eventually becomes a finished product. This is all regulated under Good Manufacturing Practice (GMP).
The January 2022 change to the regulations, for ATMPs only, allow for a hospital pharmacy to carryout additional processing operations (under the title ‘Reconstitution’, please refer to the original FOI for full details).
However, it is the Marketing Authorisation Holder (MAH) that is responsible for compliance with the contents of the eCTD, and meeting the label claim awarded by the MHRA on the basis of such. Any changes that are made by a hospital pharmacy will not have been included in the eCTD submitted by an MAH during the MAA submission process. This would create a legal nightmare in the event of any serious adverse event (SAE) occurring, with damages sought. Of course, that would pale into insignificance compared with the impact on a patient’s life.
There are, of course, further severe issues. For example, the hospital pharmacy will need to procure, transport, and store licensed medicinal products, with all the associated logistics challenges, and therefore would need to gain a successful Good Distribution Practice (GDP) inspection and award of a Wholesale Distribution Authorisation (WDA(H)) before beginning operations.
On the basis of this, I am calling for an immediate repeal of the 2022 GMDP changes, until there is an industry stakeholder consultation similar to the Falsified Medicines Directive, (FMD) 2011, where there were significant changes applied to the original EU legislation. The FMD current legislation still applies to the UK.
Given the gravity of this information, I would request that you escalate this to the MHRA Chair, Stephen Lightfoot, and the MHRA CEO, Dr June Raine. Alternatively, if you provide their email addresses, I can contact them direct, with you and David in copy.
Kind regards,
Hedley”